FDA Software as Medical Device – A Complete Guide

Software solutions are the key component of the quickly evolving healthcare industry. From smartphone apps that track your heart rate to advanced AI algorithms that help radiologists identify cancer, digital health solutions are becoming more complex.
A great invention is accompanied by great regulation and responsibility. Whether you’re a health tech startup founder or a physician with a great app idea, understanding what constitutes SaMD and how the FDA regulates it is crucial for turning your idea into a legally marketed product.
In this blog, you’ll know what FDA SaMD is, its essential aspects, and how it relates to broader medical device laws. This simplifies the process and enables you to innovate securely, confidently, and in complete compliance.
What Is FDA Software as a Medical Device?
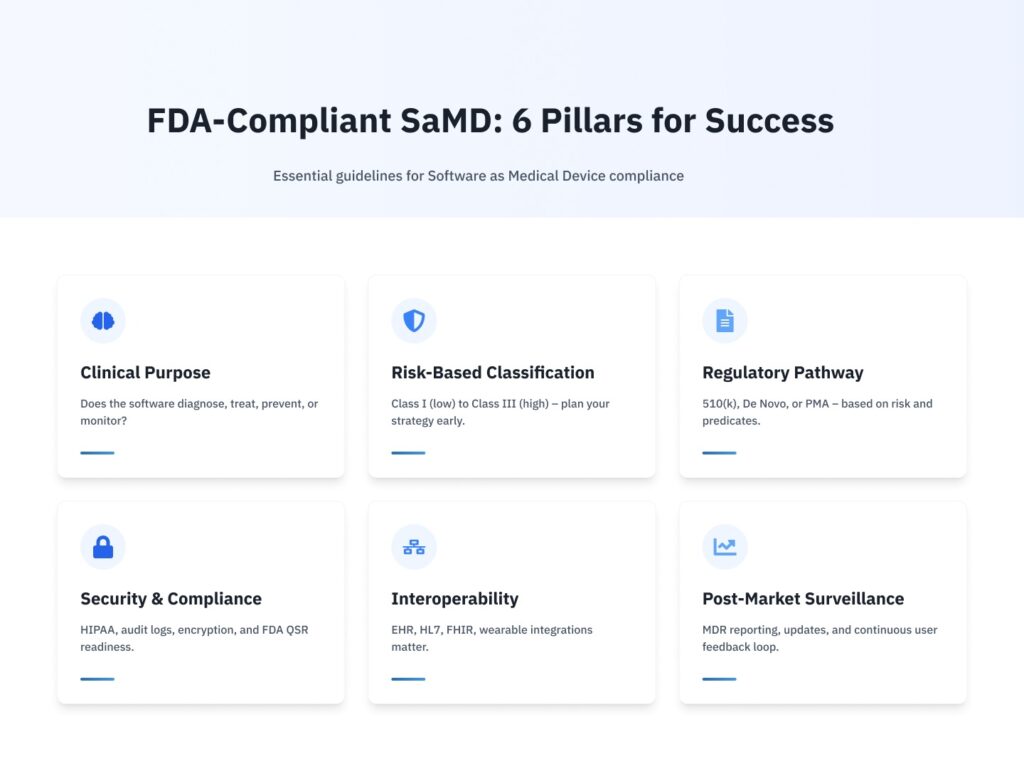
The FDA defines Software as a Medical Device as software that is intended to be used for one or more medical purposes, but does not form part of a hardware medical device. If your app diagnoses, treats, prevents, or monitors a medical condition, it may be SaMD.
- Standalone Software Functionality – Unlike software installed in a medical device, like firmware in an insulin pump, SaMD works on general-purpose computer platforms such as smartphones, tablets, and cloud servers.
- Medical Purpose – The term “medical purpose” can refer to anything from processing MRI pictures to giving clinical decision assistance or even monitoring vital signs using wearable sensors.
Key Features of SaMD
Understanding the distinguishing qualities of SaMD allows you to plan, build, and validate your solution more efficiently.
Clinical Functionality
SaMD must carry out a clinical task, like identifying disease indicators in imaging data or recommending a dosage. This suggests that you’ll need to support your claims with clinical data and thorough documentation of the intended use.
Risk-Based Classification
The FDA defines SaMD according to the danger it poses to patients.
- Low Risk (Class I) – Minimal potential harm, such as general wellness apps.
- Moderate Risk (Class II) – Assists decision-making or monitors vital indicators.
- High risk (Class III) – Directly influences crucial treatment decisions, like software that controls radiation dosing.
Recognizing your risk class early on influences your regulatory strategy and documentation plan.
Regulatory Compliance and Quality Management
The FDA’s Quality System Regulation, which covers design controls, risk management, and corrective actions, must be followed by SaMD developers. Last-minute compliance problems can be avoided by putting in place a strong quality management system from the beginning.
Interoperability and Cybersecurity
EHR systems and other devices often exchange patient data with SaMD; as a result, you need to create secure connection protocols, encrypt data, and set up threat monitoring. Maintaining interoperability reduces security threats while simultaneously enhancing the user experience.
Each of these characteristics necessitates meticulous planning, collaboration between clinical and engineering teams, and continuous dedication to both safety and innovation.
FDA Regulatory Framework for SaMD
1. Pre-Submission Engagement
Attend a Pre-Submission meeting with the FDA before starting development. This first conversation aids in defining regulatory requirements, determining which clinical trials are required, and optimizing your validation approach.
2. 510(k) Clearance: De Novo vs. PMA
Depending on your risk class and predicate devices like existing, comparable devices on the market.
- 510(k) Clearance – Show “substantial equivalence” with a predicate device typical for Class II.
- De Novo Classification – For innovative, low-to-moderate risk devices that lack a predicate.
- Premarket Approval – A rigorous clinical examination of high-risk (Class III) technologies.
3. Labeling and Directions for Use
Instructions and labels need to be simple to read and comprehend. The exact process, any contraindications, and the intended use must all be specified. When writing these sections, doctors, technical writers, and UX designers frequently need to work together to ensure accuracy and clarity.
4. Post-Market Surveillance and Reporting
Once the product is on the market, you will use the Medical Device Reporting system to report significant problems, evaluate feedback, and keep an eye out for unfavorable events. Software updates that take user input into account guarantee product compliance and consistently enhance patient results.
How SaMD Compares to Traditional Medical Devices
Although SaMD is standalone, its relationship with hardware-based medical devices is closely linked.
1. Complementary Roles
SaMD frequently improves the functionality of physical devices. For example, imaging software SaMD can give enhanced analytics for MRI equipment, allowing for earlier disease identification.
2. Integration with Device Ecosystems
Additional software is needed for many medical devices to configure, calibrate, or visualize data. Trust and usability are increased when your SaMD and hardware components communicate seamlessly.
3. Lifecycle Management Across Platforms
The life cycle expectations for hardware and software differ; whereas software changes happen quickly, equipment can remain in use for years. To provide end-to-end safety, version control, validation, and user training must be synchronized across both domains.
4. Updateability
Unlike physical devices, SaMD may be updated and improved regularly, allowing for rapid feature additions, problem fixes, and adaptation to new medical data.
Best Practices for Developing FDA-Compliant SaMD
To personalize the process, consider your growth journey to be like building a house rather than hurrying to move in. Each phase, foundation, framing, and finishing, marks important quality and regulatory milestones.
Foundation: User Needs and Requirements
Engage clinicians and end users from the beginning. Capture user stories and clinical workflows to guarantee your software solves real-world challenges.
Framing: Design Controls and Risk Management
Convert user needs into design specifications. Create risk mitigation plans in advance and carry out a hazard analysis similar to FMEA.
Finishing: Verification, Validation, and Submission
Each feature is rigorously tested to its standards. Plan and carry out clinical investigations as needed. Create a submission dossier with clear traceability from requirements to test results.
Related: Using OpenEMR to Power Rural Health & Mobile Clinics: Device, Offline, and Sync Strategies
FDA-Compliant SaMD Development with CapMinds
Bringing your Software as a Medical Device to life requires more than innovation; it demands regulatory clarity, technical precision, and seamless integration. At CapMinds, we empower health tech visionaries with end-to-end digital health services tailored for FDA-compliant SaMD development.
Our expertise includes:
- FDA Regulatory Support – Pre-submission guidance, 510(k)/De Novo/PMA strategy
- SaMD Development – Clinical functionality, risk-based classification, QMS integration
- Device Integration – Wearables, imaging software, EHRs, and other clinical systems
- Interoperability – HL7, FHIR, and API-based secure data exchange
- Cybersecurity & Compliance – HIPAA safeguards, encrypted communications, secure updates
From concept to post-market success, CapMinds accelerates your SaMD journey, securely and scalably.
Contact CapMinds today to turn your health tech idea into a market-ready, FDA-compliant solution.




